在“双碳”背景下,非CO2温室气体减排同样至关重要。本研究创新性地在450 °C下将CH4和N2O协同转化为合成气(CO和H2),提出了一种高效的N2O-甲烷干重整路径(N2O-DRM)。通过构建Gd2O3负载Ir单原子催化剂,实现了高效转化(N2O转化率99.3%,CH4转化率48.2%),同时抑制了产物的过度氧化。机理研究表明,稀土氧化钆通过缓释氧物种促进CO的选择性生成,Ir单原子则有效促进CH4活化与H2形成。
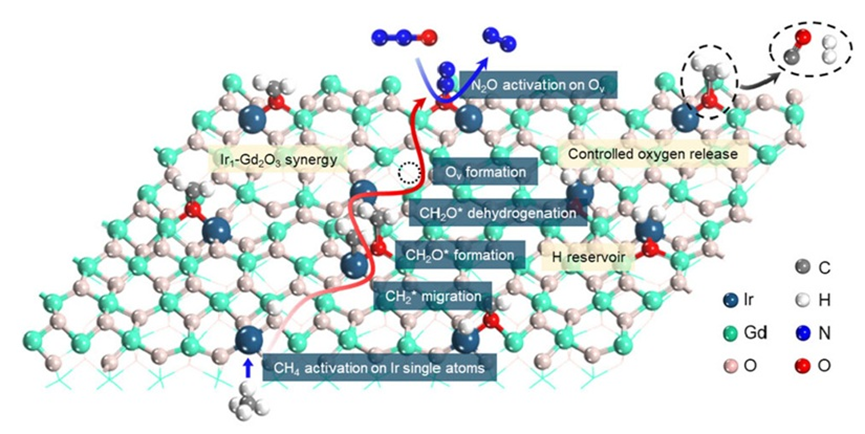
全球变暖是21世纪最严峻的环境挑战之一。其中,氧化亚氮(N2O)是第三大温室气体,其温室效应是二氧化碳的273倍,且其大气寿命超过110年。更令人担忧的是,N2O还是目前已知最主要的臭氧层破坏物质。工业生产过程中(如己二酸和己内酰胺的合成)会集中排放高浓度的N2O(体积分数高达10−40%),亟需高效治理。与此同时,甲烷(CH4)作为第二大温室气体,其温室效应是CO2的28倍,在天然气和页岩气中广泛存在,具备重要的碳资源价值。在全球“碳中和”目标下,仅控制CO2已不足以应对挑战,N2O和CH4的协同转化(Covalorization)成为碳减排和资源化利用的潜力路径。
合成气(CO + H2)是重要的基础平台化学品,可通过费托合成进一步转化为燃料或高附加值化学品。传统CO2驱动的甲烷干重整(CO2-DRM)是制备合成气的重要方法,但往往需要超过650 °C的高温,存在能耗高、反应选择性差及催化剂烧结失活等问题。受此前该团队关于N2O与丙烷干重整研究的启发(ACS Catal. 2024, 14, 13520−13530),N2O-甲烷干重整(N2O-DRM)被提出,有望以更低温度实现合成气制备,并同步削减两种强温室气体。N2O-DRM催化剂的设计需同时满足:促进CH4、N2O活化,抑制过度氧化。氧亲和力的调控被认为是影响反应路径和产物选择性的关键。此前在Fe-ZSM-5和Cu-ZSM-5体系中,调控N2O分解所产生的“单原子氧”中间体已被用于开发选择性甲烷氧化过程。
本研究设计构筑了Gd2O3负载的Ir单原子催化剂(Ir1-Gd2O3),实现了在仅450 °C下的高效N2O-DRM反应。稀土氧化钆具有独特的缓释氧物种能力,能够实现可控的活性氧释放,减少副反应。而Ir单原子位点则有效活化CH4并作为H*储库以生成H2,从而实现高效的温室气体协同转化。本研究不仅首次在热催化研究中展示了Gd2O3单原子催化体系的优异性能,也为非CO2温室气体的资源化利用提供了全新策略。
催化剂的制备与表征
作者首先制备了Ir1-Gd2O3单原子催化剂,并作为对比制备了Ir颗粒负载型催化剂(Irnano/Gd2O3)。为了进一步探究载体效应,还合成了Ir1-CeO2和Ir1-TiO2单原子催化剂。通过球差电镜与同步辐射技术,确认了目标催化剂的成功合成。XAFS和XPS测试结果显示,Ir物种以高价态的单原子形式稳定分散在载体表面,且在Gd2O3上主要呈现五配位结构。相比之下,Irnano/Gd2O3中Ir物种以金属态团簇的形式存在,体现出明显的结构差异。此外,CeO2和TiO2负载的Ir单原子催化剂也表现出稳定的Ir−O配位结构。这些结构信息为后续的DFT模拟提供了可靠依据。
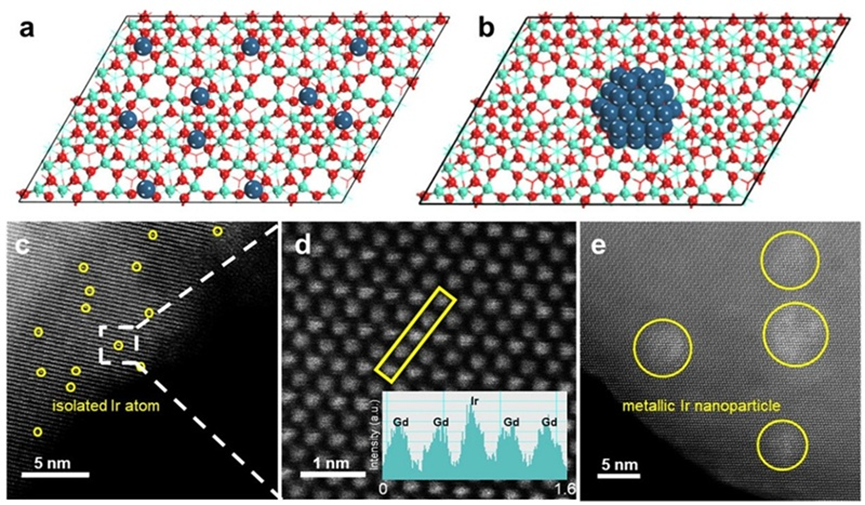
图1. (a–b) 催化剂原子结构示意图,(c–e) 球差电镜图
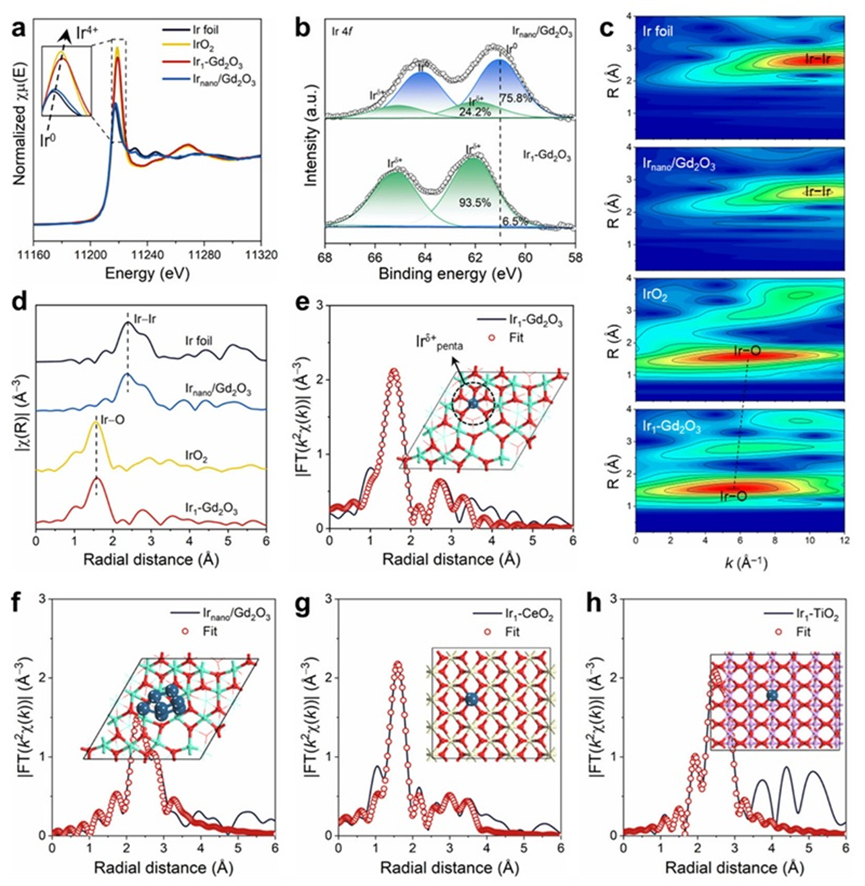
图2. (a) Ir L3-edge XANES. (b) XPS. (c) 小波转换结果. (d) EXAFS. (e−g) EXAFS拟合结果:Ir1-Gd2O3 (e), Irnano/Gd2O3 (f), Ir1-CeO2 (g), and Ir1-TiO2 (h).
催化剂性能测试
研究团队在固定床流动反应器中系统评估了所制备催化剂的N2O-CH4干重整性能。Ir1-Gd2O3在仅450 °C下就实现了99.3%的N2O转化率和48.2%的CH4转化率,CO和H2的生成速率分别为138.3 mol kgcat−1 h−1和184.9 mol kgcat−1 h−1,H2/CO比为1.34,在连续反应100小时后仍保持稳定,展现出出色的稳定性和应用潜力。相比之下,Irnano/Gd2O3中存在大量金属团簇,其CO选择性显著下降,仅为45.7%,CO生成速率为68.5 mol kgcat−1 h−1,仅为Ir1-Gd2O3的约一半,说明纳米Ir易诱导CO过度氧化。
而Ir1-CeO2尽管CH4转化率较高(56.3%),但CO选择性仅为27.1%,说明CeO2强氧亲和性倾向于生成CO2。Ir1-TiO2的整体活性最低,进一步表明TiO2不利于CH4/N2O活化。此外,Ir1-Gd2O3催化剂在使用不同氧化剂(如O2或CO2)时,表现出显著差异:O2虽可提高CH4转化率,但CO选择性大幅下降;而CO2则几乎不发生反应。该结果强调了Gd2O3载体对于N2O提供的活性氧物种的调控作用,能够有效避免产物过度氧化,保障CO和H2的生成。
综合而言,Ir1-Gd2O3在低温、高选择性、高稳定性等方面均优于其他对比样品,是实现N2O与CH4协同高效转化为合成气的优质候选。
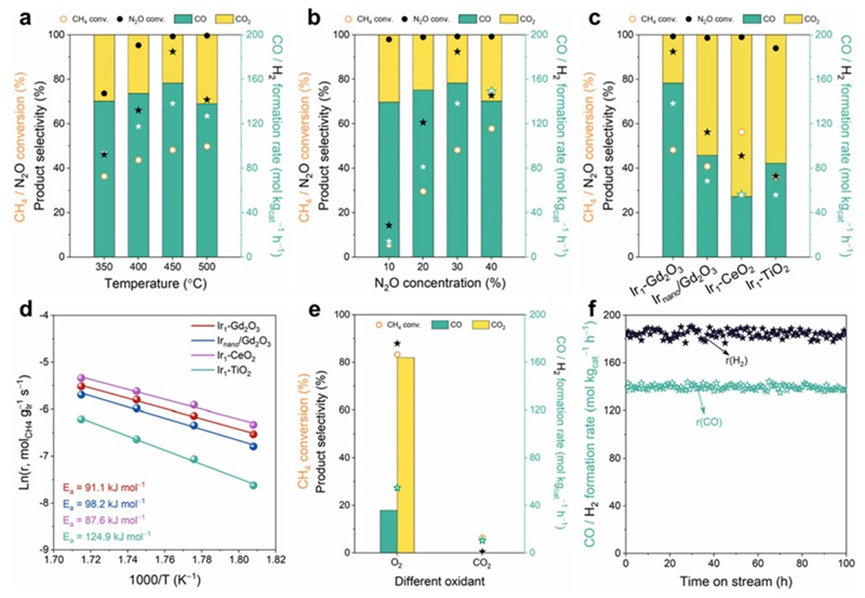
图3. N2O-DRM活性测试结果. (a) 不同反应温度. (b) 不同CH4/N2O比例. (c) 不同催化剂. (d) 反应动力学测试. (e) 不同氧化剂. (f) 稳定性测试.
反应机理解析
通过近常压X射线光电子能谱(NAP-XPS)、程序升温还原(H2-TPR)、原位红外(DRIFTS)和密度泛函理论(DFT)计算,作者详尽地揭示了Ir1-Gd2O3催化剂在N2O-DRM反应中合成气生成的反应机理。研究表明,Gd2O3是一种优异的催化剂载体:相比CeO2易造成产物过度氧化、TiO2则难以活化CH4,Gd2O3在二者之间实现了良好平衡,既能有效抑制过度氧化,又促进CH4活化。此外,相较于单原子Ir(Ir SAs),Ir纳米颗粒(Ir NPs)表现出更差的选择性。这是因为N2O不仅可在Gd2O3表面的氧空位上被活化,也可在Ir NPs上直接解离,生成大量局部活性氧(O*)物种,这些O*极易与中间产物或CO发生反应,导致过度氧化生成CO2,显著降低CO选择性。而在Ir1-Gd2O3单原子催化剂中,活性位点更倾向于将N2O活化过程限制在Gd2O3的氧空位上,并且由于其强氧亲和力实现了O*的缓释,有效降低了副反应的发生概率。
在N2O-CH4干重整(N2O-DRM)反应过程中,Gd2O3作为载体与Ir单原子位点形成了协同作用。其中,N2O的活化主要发生在Gd2O3表面的氧空位(Ov)上,Ov具有对电负性氧原子的良好亲和性,使N2O分子在此高效解离,生成单原子氧(O*)物种,并释放N2。相比之下,CH4分子的活化则由高度分散的Irδ+单原子位点完成,Irδ+位点可以稳定吸附CH4并促进其C–H键断裂,形成CHx*中间体(如CH3*、CH2*等)。这一过程是整个反应的速率控制步骤。随后,CHx*中间体会进一步迁移至Gd2O3表面,与O物种反应生成CO或CO2前驱体。值得注意的是,由于Ir为单原子状态,其表面并不富集过量O,从而有效抑制了中间体的深度氧化,使得CO得以顺利生成并脱附。与此同时,活性H物种有向Ir单原子位点聚集的趋势,形成H储库,从而显著提升H–H偶联概率,促进H2的生成。
整体而言,Ir1-Gd2O3通过“位点分工”与“氧物种缓释”策略,实现了N2O的高效活化、CH4的精准裂解与中间体的选择性氧化,使该催化剂在低温下兼具高转化率与高CO/H2产率,展现出在温和条件下协同利用温室气体的巨大潜力。
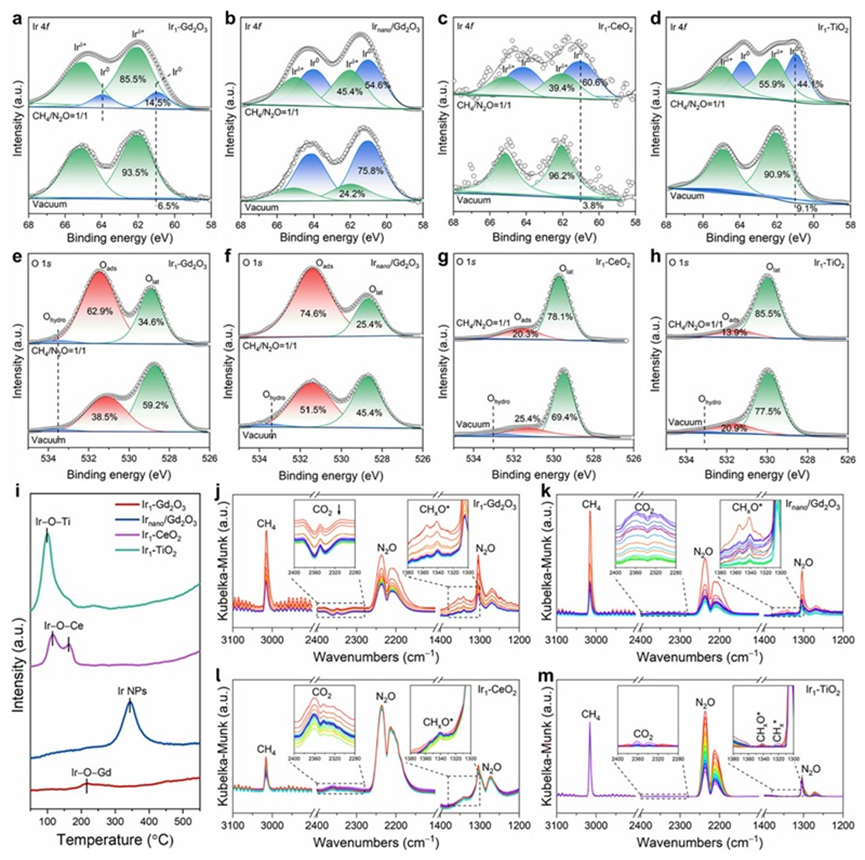
图4. 反应机理实验表征. (a–h) NAP-XPS谱图(CH4/N2O = 1/1,450 °C). (i) H2-TPR. (j–m) 反应原位红外谱图
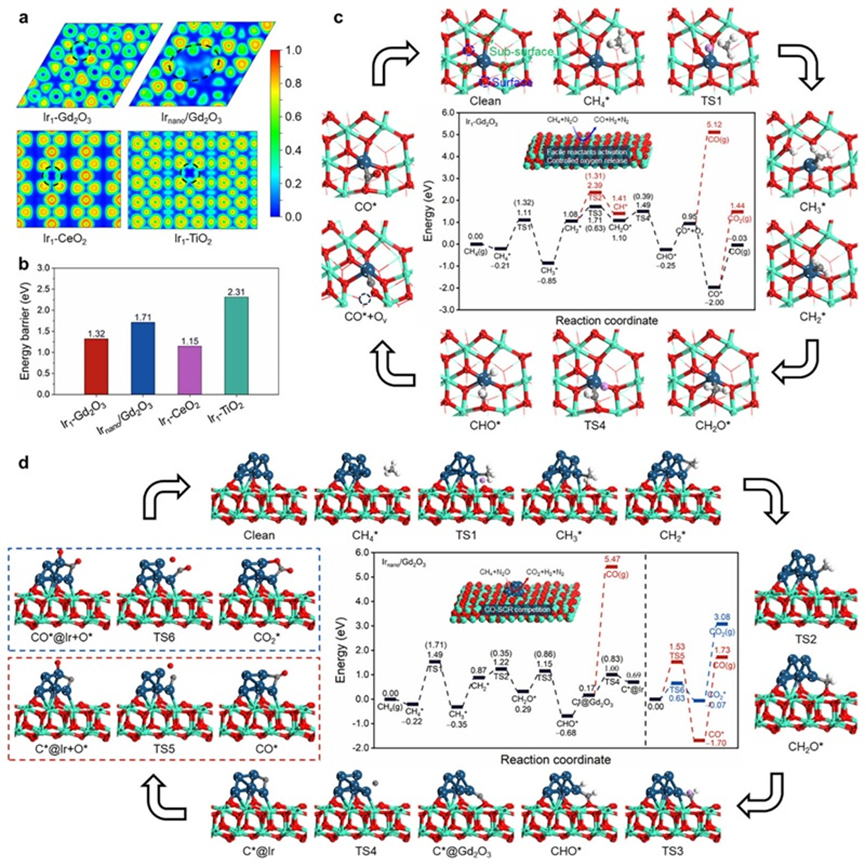
图5. DFT计算. (a) ELF结果. (b) CH4活化能垒比较. (c) Ir1-Gd2O3反应路径图. (d) Irnano/Gd2O3反应路径图
本研究通过设计基于Gd2O3负载的五配位铱单原子催化剂(Ir1-Gd2O3),成功实现了催化活性位点与载体功能的高效协同。实验表明,在450 °C下,该催化剂在N2O干法重整甲烷(N2O-DRM)反应中展现出优异的催化性能,N2O转化率高达99.3%,同时实现了高选择性的合成气生成。机理研究揭示,Ir单原子位点主要负责甲烷的活化与裂解,而Gd2O3载体则有效稳定由N2O产生的活性氧物种,促进了氧化还原过程的合理调控,抑制了过氧化副反应,最终实现了高效且稳定的合成气产率。该研究不仅为非CO2温室气体的高效资源化利用提供了创新策略,也为多相催化剂设计开辟了新的思路。未来,通过进一步优化催化剂结构与反应条件,有望提升催化剂的工业适用性与稳定性。同时,单原子催化、载体调控以及双功能活性位点设计理念可推广应用于其他温室气体转化和绿色化工过程,助力实现可持续发展目标。

